DeepCAGE: Incorporating Transcription Factors in Genome-Wide Prediction of Chromatin Accessibility
Abstract
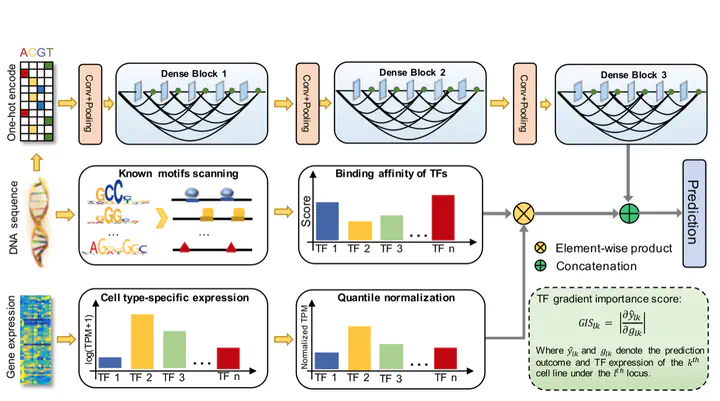
Although computational approaches have been complementing high-throughput biological experiments for the identification of functional regions in the human genome, it remains a great challenge to systematically decipher interactions between transcription factors (TFs) and regulatory elements to achieve interpretable annotations of chromatin accessibility across diverse cellular contexts. To solve this problem, we propose DeepCAGE, a deep learning framework that integrates sequence information and binding statuses of TFs, for the accurate prediction of chromatin accessible regions at a genome-wide scale in a variety of cell types. DeepCAGE takes advantage of a densely connected deep convolutional neural network architecture to automatically learn sequence signatures of known chromatin accessible regions and then incorporates such features with expression levels and binding activities of human core TFs to predict novel chromatin accessible regions. In a series of systematic comparisons with existing methods, DeepCAGE exhibits superior performance in not only the classification but also the regression of chromatin accessibility signals. In a detailed analysis of TF activities, DeepCAGE successfully extracts novel binding motifs and measures the contribution of a TF to the regulation with respect to a specific locus in a certain cell type. When applied to whole-genome sequencing data analysis, our method successfully prioritizes putative deleterious variants underlying a human complex trait and thus provides insights into the understanding of disease-associated genetic variants. DeepCAGE can be downloaded from https://github.com/kimmo1019/DeepCAGE.