Abstract
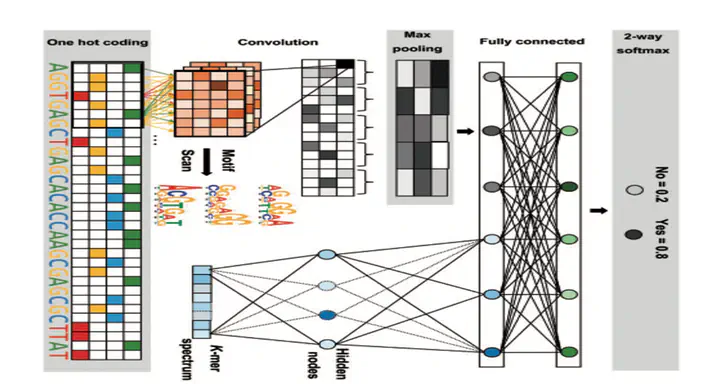
A majority of known genetic variants associated with human-inherited diseases lie in non-coding regions that lack adequate interpretation, making it indispensable to systematically discover functional sites at the whole genome level and precisely decipher their implications in a comprehensive manner. Although computational approaches have been complementing high-throughput biological experiments towards the annotation of the human genome, it still remains a big challenge to accurately annotate regulatory elements in the context of a specific cell type via automatic learning of the DNA sequence code from large-scale sequencing data. Indeed, the development of an accurate and interpretable model to learn the DNA sequence signature and further enable the identification of causative genetic variants has become essential in both genomic and genetic studies. We proposed Deopen, a hybrid framework mainly based on a deep convolutional neural network, to automatically learn the regulatory code of DNA sequences and predict chromatin accessibility. In a series of comparison with existing methods, we show the superior performance of our model in not only the classification of accessible regions against background sequences sampled at random, but also the regression of DNase-seq signals. Besides, we further visualize the convolutional kernels and show the match of identified sequence signatures and known motifs. We finally demonstrate the sensitivity of our model in finding causative noncoding variants in the analysis of a breast cancer dataset. We expect to see wide applications of Deopen with either public or in-house chromatin accessibility data in the annotation of the human genome and the identification of non-coding variants associated with diseases
Qiao Liu
Assistant Professor of Biostatistics
My research interests include generative AI, high-dimensional data analysis, and computational biology.